How to analyse multi-omics data?
Last updated on:4 months ago
Multi-omics data refers to the integration and analysis of data from multiple different “omics” fields, such as genomics, transcriptomics, proteomics, and metabolomics.
Multi-omics data
Introduction/What is multi-omics data
Multi-omics data refers to the combined data from various biological “omes”, such as genome, transcriptome, proteome, epigenome, and metabolome. It is an approach that integrates different datasets to gain a more holistic understanding of biological systems, diseases, and potential therapeutic interventions.
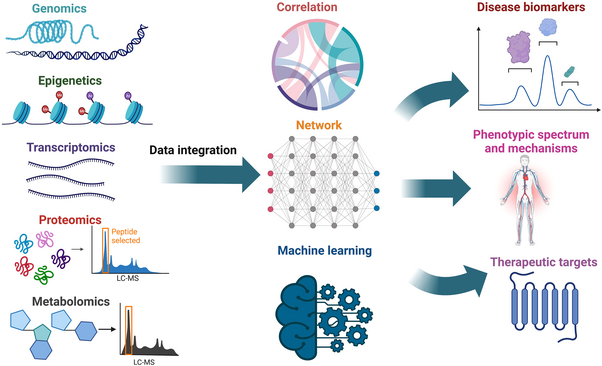
What are the “omes” in multi-omics?
- Genome: the complete set of DNA instructions for an organism.
- Transcriptome: all the RNA molecules in a cell or organism.
- Proteome: all the proteins in a cell or organism.
- Epigenome: chemical modifications to DNA and histones that affect gene expression.
- Metabolome: all the small molecules (metabolites) in a cell or organism.
Multiomics, multi-omics, integrative omics, phenomics, or pan-omics is a biological analysis approach in which the data consists of multiple “homes”.
Chinese introduction
多组学(Multi-Omics) 是指结合多种“组学”技术,从不同层次和维度全面解析生物系统的复杂性。传统的单一组学研究通常关注基因组、转录组、蛋白质组、代谢组等某一特定层面的数据,而多组学则通过整合这些多层次的数据,提供一个更加全面和系统化的视角来理解生物过程。
- 基因组学(Genomics):研究生物体的全部遗传信息,即DNA序列及其变异。
- 转录组学(Transcriptomics):分析细胞或组织中所有RNA分子的表达水平,包括mRNA、lncRNA、miRNA等。
- 蛋白质组学(Proteomics):研究细胞或组织中的所有蛋白质种类、结构和功能。
- 代谢组学(Metabolomics):检测和定量细胞或生物体内所有小分子代谢物的组成和变化。
- 表观基因组学(Epigenomics):研究基因组上的化学修饰(如甲基化、乙酰化等)及其对基因表达的影响。
- 其他组学:还包括微生物组学(Microbiomics)、脂质组学(Lipidomics)、糖组学(Glycomics)等。
多组学的核心在于将这些不同层次的数据进行整合分析,以揭示复杂的生物网络和相互作用机制,从而更好地理解生命过程、疾病发生发展以及药物反应等现象
Protein components
Protein data is usually saved in “.pdb” file format. To describe a protein, we can use a sequence of amino acids, secondary structure (SS8), solvent accessible surface area (SASA), function keywords, and structure coordinates.
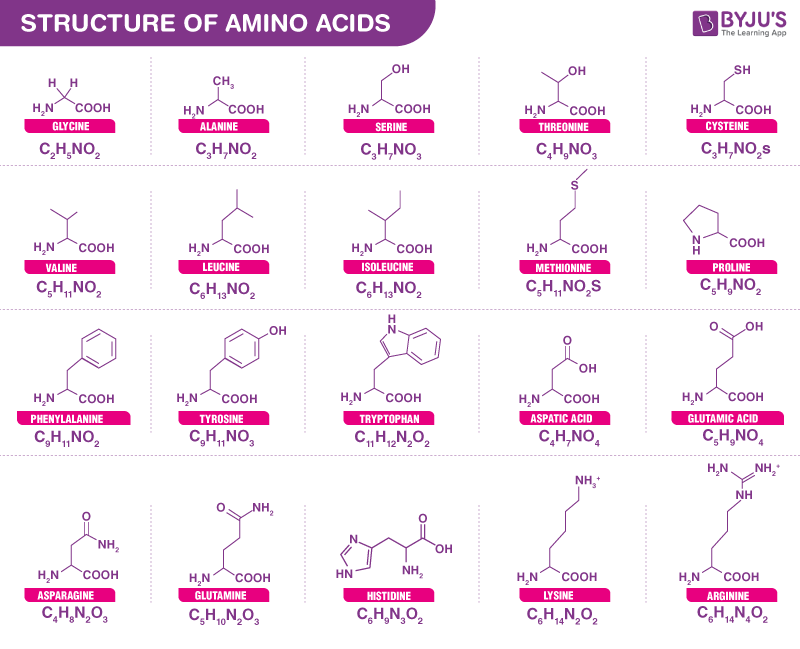
Amino acids
The fundamental component of protein is amino acids, which can be represented by code: A (alanine), R (arginine), etc.
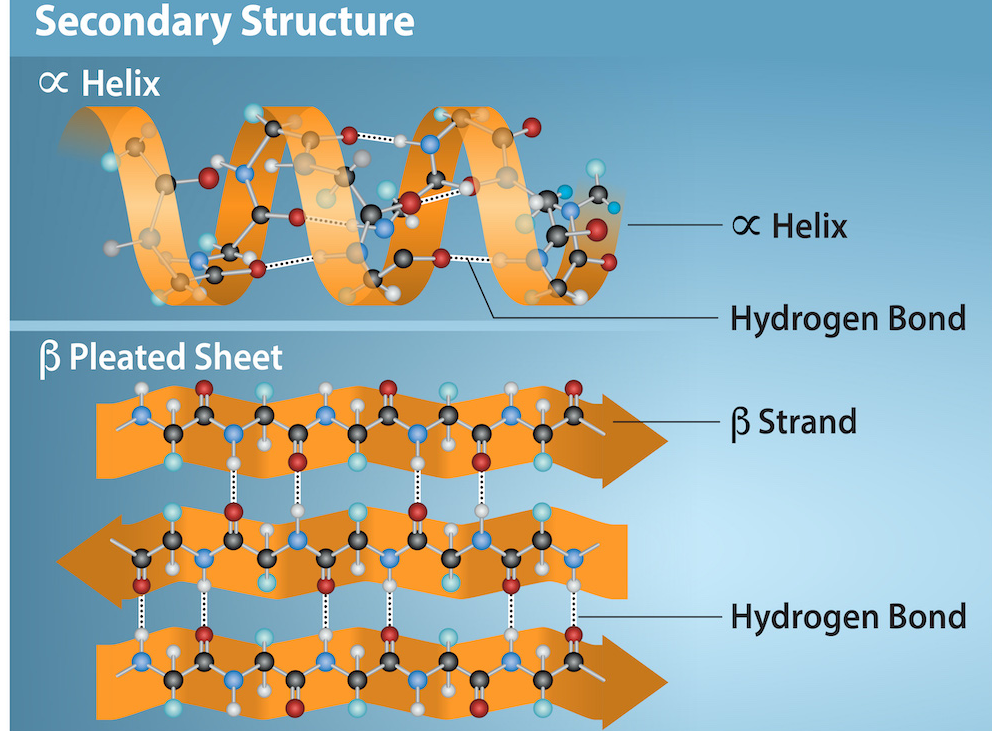
Secondary structure
The secondary structure of a protein refers to the local folding patterns of the polypeptide backbone, specifically how the amino acids are arranged in space, and is primarily driven by hydrogen bonds.
The secondary structure of a protein, classified into a 8-state system (SS8), describes the local folding patterns of the polypeptide backbone. These patterns are defined by the hydrogen bonds between the backbone atoms and are crucial for understanding protein function and structure. The 8-state secondary structure elements include alpha-helix (H), 310-helix (G), pi-helix (I), beta-strand (E), beta-bridge (B), turn (T), bend (S), and coil (C).
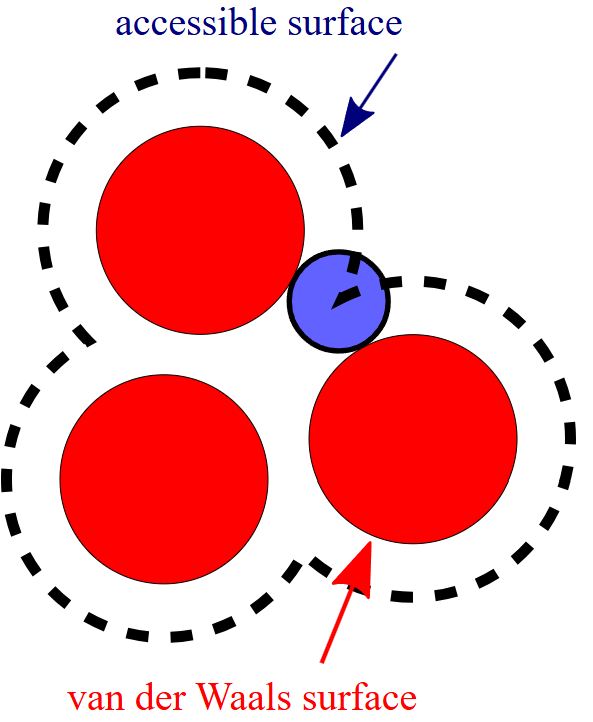
Solvent accessible surface area
Solven-accessible surface area (SASA) refers to the surface area of a molecule, typically protein, that is exposed to and can interact with a solvent. It’s a key structural property because it determines which parts of the molecule are accessible to the surrounding environment and thus influences interactions.
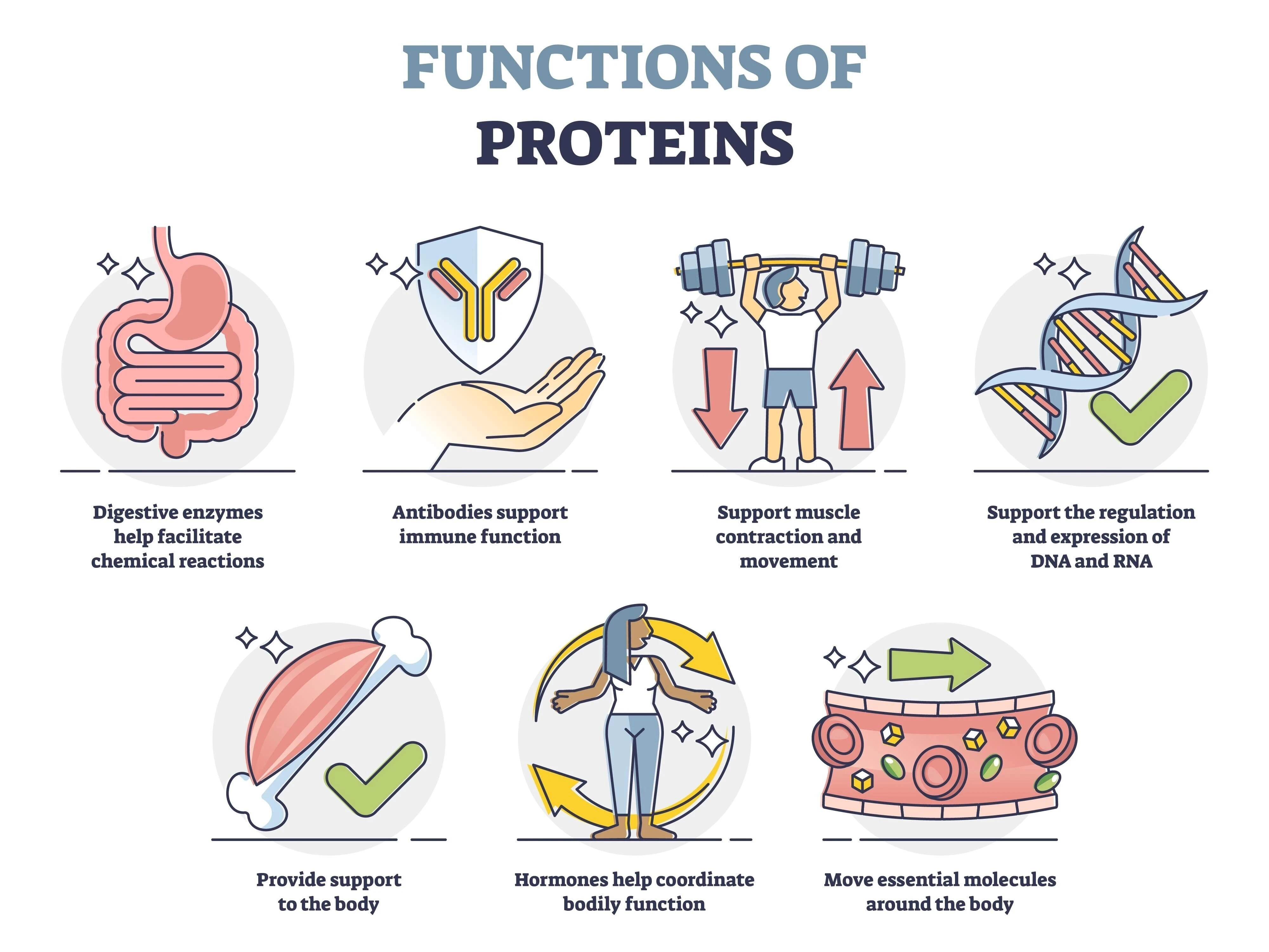
Protein functions
Proteins have diverse and crucial functions in living organisms. They act as structural components, regulatory molecules, and functional catalysts, playing a role in everything from building tissues to facilitating chemical reactions and transporting materials.
Structure coordinates
E.g., using Cartesian coordinates (X, Y, Z for all atoms) can represent the absolute position of atoms in space. At the same time, this is a commonly used representation of protein structures.
Codes
Protein data type example:
class ESMProtein(object):
# Tracks
sequence: str | None = None
secondary_structure: str | None = None
sasa: list[float | None] | None = None
function_annotations: list[FunctionAnnotation] | None = None
coordinates: torch.Tensor | None = NoneApplications
Select the protein-of-interest
Revise the application of the advanced AI method Alphafold2 to a specific task, namely protein activation, with a targeted objective of conducting clinical experiments using living mice.
aaRS evolution by machine learning:
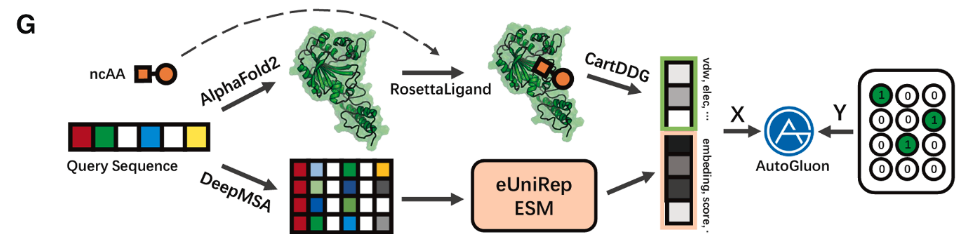
Protein structure and function prediction
Adapt AlphaFold2 for predicting the structure of conduction domain proteins.
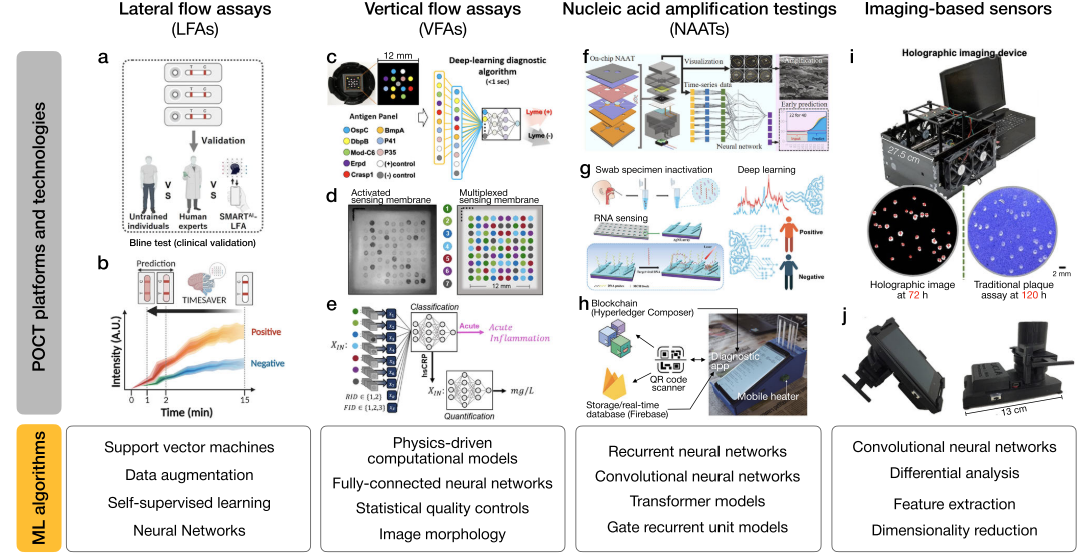
Point-of-care platforms
Integrate machine learning with clinical imaging devices and expert knowledge from flow assays to enhance specific medical tasks, such as nucleic acid amplification.
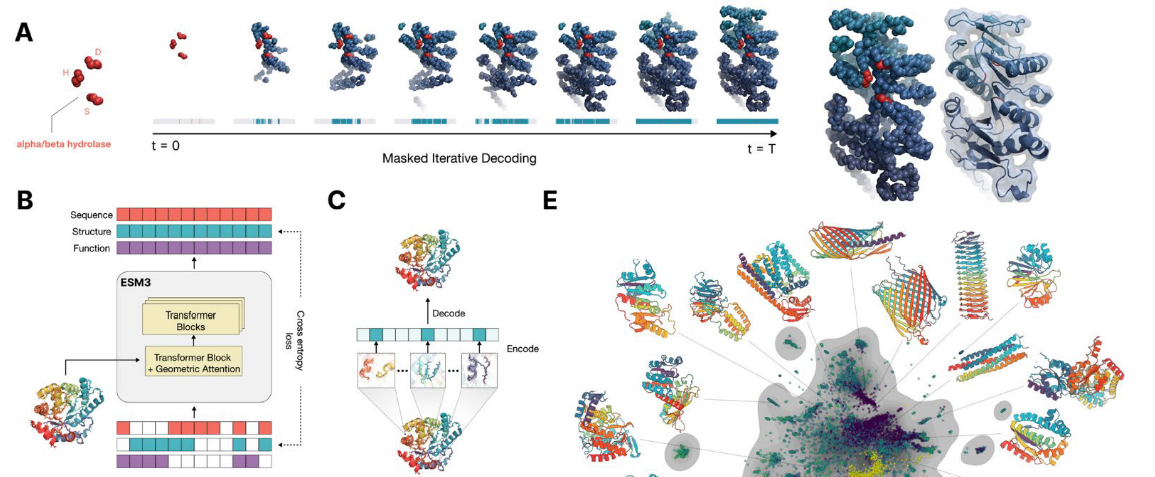
Evolution simulation by protein analysis
Utilise large language models to infer and reason about protein decoding sequences, structures, and functions.
ESM3 is a generative masked language model that allows you to provide prompts with partial sequences, structures, and function keywords. You can iteratively sample the masked positions until all positions are revealed.
Codes
Protein (e.g., Carbonic Anhydrase) generation by giving partial sequence:
# Generate a completion for a partial Carbonic Anhydrase (2vvb)
prompt = "___________________________________________________DQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPP___________________________________________________________"
protein = ESMProtein(sequence=prompt)
# Generate the sequence, then the structure. This will iteratively unmask the sequence track.
protein = model.generate(protein, GenerationConfig(track="sequence", num_steps=8, temperature=0.7))
# We can show the predicted structure for the generated sequence.
protein = model.generate(protein, GenerationConfig(track="structure", num_steps=8))
protein.to_pdb("./generation.pdb")Masked the sequence of a protein class, the sequence can be re-generated by the ESM3.
# Then we can do a round trip design by inverse folding the sequence and recomputing the structure
protein.sequence = None
protein = model.generate(protein, GenerationConfig(track="sequence", num_steps=8))
protein.coordinates = None
protein = model.generate(protein, GenerationConfig(track="structure", num_steps=8))
protein.to_pdb("./round_tripped.pdb")Encode and decode the sub-data:
# encode
if input.sequence is not None:
sequence_tokens = encoding.tokenize_sequence(
input.sequence, self. tokenisers.sequence, add_special_tokens=True
)
if input.secondary_structure is not None:
secondary_structure_tokens = encoding.tokenize_secondary_structure(
input.secondary_structure,
self. tokenisers.secondary_structure,
add_special_tokens=True,
)
if input.sasa is not None:
sasa_tokens = encoding.tokenize_sasa(
input.sasa, self. tokenisers.sasa, add_special_tokens=True
)
return ESMProteinTensor(
sequence=sequence_tokens,
structure=structure_tokens,
secondary_structure=secondary_structure_tokens,
sasa=sasa_tokens,
function=function_tokens,
residue_annotations=residue_annotation_tokens,
coordinates=coordinates,
).to(next(self.parameters()).device)
# decode
decode_protein_tensor(
input=input,
tokenizers=self. tokenisers,
structure_token_decoder=self.get_structure_decoder(),
function_token_decoder=self.get_function_decoder(),
)Learning process, forward:
def forward(
self,
*,
sequence_tokens: torch.Tensor | None = None,
structure_tokens: torch.Tensor | None = None,
ss8_tokens: torch.Tensor | None = None,
sasa_tokens: torch.Tensor | None = None,
function_tokens: torch.Tensor | None = None,
residue_annotation_tokens: torch.Tensor | None = None,
average_plddt: torch.Tensor | None = None,
per_res_plddt: torch.Tensor | None = None,
structure_coords: torch.Tensor | None = None,
chain_id: torch.Tensor | None = None,
sequence_id: torch.Tensor | None = None,
) -> ESMOutput:
"""
Performs forward pass through the ESM3 model. Check utils to see how to tokenise inputs from raw data.
Args:
sequence_tokens (torch.Tensor, optional): The amino acid tokens.
structure_tokens (torch.Tensor, optional): The structure tokens.
ss8_tokens (torch.Tensor, optional): The secondary structure tokens.
sasa_tokens (torch.Tensor, optional): The solvent accessible surface area tokens.
function_tokens (torch.Tensor, optional): The function tokens.
residue_annotation_tokens (torch.Tensor, optional): The residue annotation tokens.
average_plddt (torch.Tensor, optional): The average plddt across the entire sequence.
per_res_plddt (torch.Tensor, optional): The per residue plddt, if you want to specify exact plddts, use this; otherwise, use average_plddt.
structure_coords (torch.Tensor, optional): The structure coordinates, in the form of (B, L, 3, 3).
chain_id (torch.Tensor, optional): The chain ID
sequence_id (torch.Tensor, optional): The sequence ID.
Returns:
ESMOutput: The output of the ESM3 model.
Raises:
ValueError: If at least one of the inputs is None.
"""
# Reasonable defaults:
try:
L, device = next(
(x.shape[1], x.device)
for x in [
sequence_tokens,
structure_tokens,
ss8_tokens,
sasa_tokens,
structure_coords,
function_tokens,
residue_annotation_tokens,
]
if x is not None
)
except StopIteration:
raise ValueError("At least one of the inputs must be non-None")
t = self.tokenizers
defaults = lambda x, tok: (
torch.full((1, L), tok, dtype=torch.long, device=device) if x is None else x
)
sequence_tokens = defaults(sequence_tokens, t.sequence.mask_token_id)
ss8_tokens = defaults(ss8_tokens, C.SS8_PAD_TOKEN)
sasa_tokens = defaults(sasa_tokens, C.SASA_PAD_TOKEN)
average_plddt = defaults(average_plddt, 1).float()
per_res_plddt = defaults(per_res_plddt, 0).float()
chain_id = defaults(chain_id, 0)
if residue_annotation_tokens is None:
residue_annotation_tokens = torch.full(
(1, L, 16), C.RESIDUE_PAD_TOKEN, dtype=torch.long, device=device
)
if function_tokens is None:
function_tokens = torch.full(
(1, L, 8), C.INTERPRO_PAD_TOKEN, dtype=torch.long, device=device
)
if structure_coords is None:
structure_coords = torch.full(
(1, L, 3, 3), float("nan"), dtype=torch.float, device=device
)
structure_coords = structure_coords[
..., :3, :
] # In case we pass in an atom14 or atom37 repr
affine, affine_mask = build_affine3d_from_coordinates(structure_coords)
structure_tokens = defaults(structure_tokens, C.STRUCTURE_MASK_TOKEN)
assert structure_tokens is not None
structure_tokens = (
structure_tokens.masked_fill(structure_tokens == -1, C.STRUCTURE_MASK_TOKEN)
.masked_fill(sequence_tokens == C.SEQUENCE_BOS_TOKEN, C.STRUCTURE_BOS_TOKEN)
.masked_fill(sequence_tokens == C.SEQUENCE_PAD_TOKEN, C.STRUCTURE_PAD_TOKEN)
.masked_fill(sequence_tokens == C.SEQUENCE_EOS_TOKEN, C.STRUCTURE_EOS_TOKEN)
.masked_fill(
sequence_tokens == C.SEQUENCE_CHAINBREAK_TOKEN,
C.STRUCTURE_CHAINBREAK_TOKEN,
)
)
x = self.encoder(
sequence_tokens,
structure_tokens,
average_plddt,
per_res_plddt,
ss8_tokens,
sasa_tokens,
function_tokens,
residue_annotation_tokens,
)
x, embedding, _ = self.transformer(
x, sequence_id, affine, affine_mask, chain_id
)
return self.output_heads(x, embedding)Turn the prediction to regression tasks:
self.output_heads = OutputHeads(d_model) # e.g., d_model=1536 for ESM3_sm_open_v0
class OutputHeads(nn.Module):
def __init__(self, d_model: int):
super().__init__()
self.sequence_head = RegressionHead(d_model, 64)
self.structure_head = RegressionHead(d_model, 4096)
self.ss8_head = RegressionHead(d_model, 8 + 3)
self.sasa_head = RegressionHead(d_model, 16 + 3)
self.function_head = RegressionHead(d_model, 260 * 8)
self.residue_head = RegressionHead(d_model, 1478)
def forward(self, x: torch.Tensor, embed: torch.Tensor) -> ESMOutput:
sequence_logits = self.sequence_head(x)
structure_logits = self.structure_head(x)
secondary_structure_logits = self.ss8_head(x)
sasa_logits = self.sasa_head(x)
function_logits = self.function_head(x)
function_logits = einops.rearrange(function_logits, "... (k v) -> ... k v", k=8)
residue_logits = self.residue_head(x)
return ESMOutput(
sequence_logits=sequence_logits,
structure_logits=structure_logits,
secondary_structure_logits=secondary_structure_logits,
sasa_logits=sasa_logits,
function_logits=function_logits,
residue_logits=residue_logits,
embeddings=embed,
)Reference
- Wang, X., Liu, Y., Wang, Z., Zeng, X., Ngai, W.S.C., Wang, J., Zhang, H., Xie, X., Zhu, R., Fan, X. and Wang, C., 2025. Machine-learning-assisted universal protein activation in living mice. Cell.
- 再谈多组学(multi-omics)
- Wang, X., Liu, Y., Wang, Z., Zeng, X., Ngai, W.S.C., Wang, J., Zhang, H., Xie, X., Zhu, R., Fan, X. and Wang, C., 2025. Machine-learning-assisted universal protein activation in living mice. Cell.
- Han, G.R., Goncharov, A., Eryilmaz, M., Ye, S., Palanisamy, B., Ghosh, R., Lisi, F., Rogers, E., Guzman, D., Yigci, D. and Tasoglu, S., 2025. Machine learning in point-of-care testing: innovations, challenges, and opportunities. Nature Communications, 16(1), p.3165.
- Hayes, T., Rao, R., Akin, H., Sofroniew, N.J., Oktay, D., Lin, Z., Verkuil, R., Tran, V.Q., Deaton, J., Wiggert, M. and Badkundri, R., 2025. Simulating 500 million years of evolution with a language model. Science, p.eads0018.
- esm
- Multiomics
- Understanding Internal Coordinates in Protein Structure Representation
- Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A. and Bridgland, A., 2021. Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), pp.583-589.
本博客所有文章除特别声明外,均采用 CC BY-SA 4.0 协议 ,转载请注明出处!